
Ibogaine Is Not What You Think
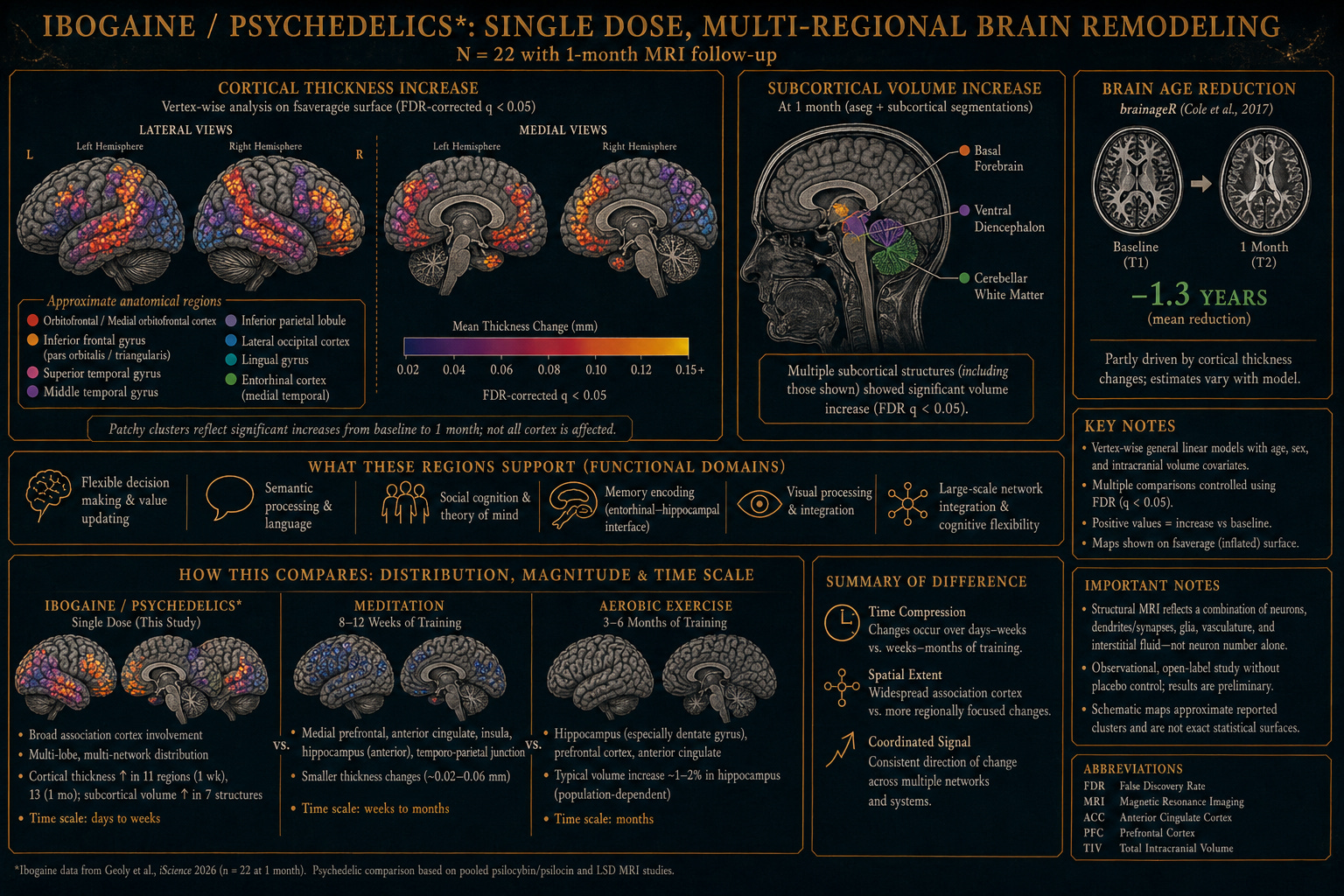
A single experience was followed by thicker cortex, expanded subcortical volume, and biological brain age 1.3 years younger.


What if the brain has a way of remodeling itself we’ve been almost entirely wrong about for fifty years?
A team at Stanford in association with the Palo Alto VA gave thirty US Navy SEAL special operations veterans a single oral dose of ibogaine, paired with intravenous magnesium for cardiac stability. Each veteran was carrying traumatic brain injuries from blast exposure, plus various combinations of PTSD, depression, anxiety, and functional disability that conventional treatment had not been able to resolve. The clinical results, published in Nature Medicine in early 2024, showed shifts in those symptoms at magnitudes the existing TBI literature simply does not contain.1
This year, the structural follow-up appeared. Same cohort, same protocol — but this time the brains themselves were measured, in detail, before and after. The paper, in iScience, describes something even more striking than the clinical findings.2 Cortical thickness rose in eleven regions one week after the dose, and in thirteen regions a month out. Subcortical volume expanded in seven anatomical structures, including cerebellar white matter, basal forebrain, and ventral diencephalon. And by the standard machine-learning metric of biological brain age3 — trained on tens of thousands of structural MRIs — the average brain in the cohort had become 1.3 years younger at one month, relative to where it started.
A single dose, in other words, produced sustained, multi-regional structural change in the direction of healthier and younger neural tissue — in a population that conventional medicine considers among the hardest to help.
The Stanford team — led by Dr. Maheen Adamson, building on work from the Stanford Brain Stimulation Lab founded by Nolan Williams before his death in October 2025 — is more precise in its implication. If a single intervention can induce coordinated, system-level remodeling that durably persists, then the question is not just what the molecule does, but what class of intervention it represents — and what assumptions about brain change it forces the field to revise.
What 1.3 years means, in context
It’s worth pausing on that number, because it sits in a strange place in the literature. A brain-age reduction of more than a year, from a single pharmacologic dose, sustained at one month, is a rare result — the Stanford authors themselves place it in that rare context. Among published interventions that have successfully improved predicted brain age, the closest comparators are surgical treatment of mesial temporal lobe epilepsy (about five years over two years post-surgery), electroconvulsive therapy in some depression cohorts, sustained exercise training (measured in isolation), testosterone replacement, the postpartum recovery period, and a single small ibuprofen study (about 1.1 years). Most pharmacologic interventions do not move this metric at all.
The honest caveats matter and the authors name them. T1-weighted MRI is sensitive to nonstructural variation — tissue water content, signal-to-noise differences. The brainageR algorithm like many learning models, in their own words, “in some ways a black box,” and increases in cortical thickness in this cohort likely contributed mechanically to the age estimate. The trial was small (n = 22 at one month), open-label, with no placebo arm, and the population — special operations veterans selected partly on physiological capacity — had brains already younger than their chronological age at baseline. The authors are careful to call the findings “compatible with increased structural neuroplasticity” rather than diagnostic of it.
Even with all of those caveats acknowledged, the finding is striking. It is consistent across multiple structural measures — cortical thickness, subcortical volume, and the brain-age estimate. It is sustained. And it is the first published structural-MRI evidence in humans that a single dose of ibogaine, in this protocol, is associated with brain change in the direction of more, and younger, neural tissue.
What the molecule does
Ibogaine is unusual at the receptor level, and the unusualness is part of what makes the structural data interpretable and not so mysterious. The molecule engages NMDA glutamate receptors, kappa-opioid receptors, sigma-2 receptors, and nicotinic acetylcholine receptors, with weaker but real serotonergic activity through both the parent compound and its long-half-life metabolite, noribogaine.4 In midbrain dopaminergic neurons, it induces expression of glial cell line–derived neurotrophic factor (GDNF) and of brain-derived neurotrophic factor (BDNF)5 at levels and durations that are not characteristic of compounds the field has historically classified as drugs.
GDNF and BDNF are the signals the nervous system uses to repair, remodel, and stabilize neural circuitry. They do not appear in significant quantities in response to most molecules in our pharmacopeia. They are upregulated by many domains I write about in Adaptive Resilience, like exercise, certain forms of fasting, and sleep — and they are upregulated by the compounds the brain appears to use, when it has access to them, for structural reorganization.
The Stanford structural finding is interpretable in exactly this frame. Cortical thickening, subcortical volume expansion, and brain-age reduction of the kind reported are what one would expect from sustained neurotrophic-factor upregulation acting on neural tissue, in a system that has been holding itself rigid against trauma. The molecule is not analgesic, not sedative, not stimulating in any conventional pharmacological sense. It is functioning closer to an instruction — a transient, time-limited disruption that the system reorganizes around, with the reorganization visible at the level of cortical anatomy a month later.
The substrate question
There is one part of the ibogaine story that bears on why the Stanford safety profile looked different from what one might have expected from the historical record.
Ibogaine prolongs the QT interval, and the historical record of cardiac events — including deaths — is real. What is striking, looking at where that record came from, is the population it was drawn from: people with long histories of polysubstance use, treated in unregulated clinics, often without continuous cardiac monitoring, often without protective electrolyte support, frequently with concurrent QT-prolonging medications and undiagnosed cardiac comorbidities.
This is not the population from which one extracts a clean estimate of intrinsic compound risk. It is the population in which several substrate-level variables — mitochondrial reserve, electrolyte balance, cardiac tissue integrity, autonomic regulation — were already severely compromised before any new molecule entered the picture.
Adaptive Resilience treats substrate state as load-bearing. A system with intact energetic reserve absorbs an acute load of a given size. A system already running near its ceiling does not. The historical ibogaine record is what one would expect when a compound that produces an acute energetic demand meets a substrate that was already failing. The Stanford record is what one would expect when the same compound meets a substrate that is intact, with magnesium added at the moment of peak cardiac demand. These are not contradictory datasets. They are samples from opposite ends of a distribution the conversation has not yet fully named.
The shape of recovery
The 1.3-year brain-age reduction is what makes the mycologist Paul Stamets’ framing (psilocybin may be a kind of nutrient with many benefits) more than evocative.6 But it is not, by itself, the most important thing the data is suggesting. The most important thing is the shape of what that healing turned out to be.
The damage that combat-related TBI and PTSD produce is not only symptomatic. The phenotype includes hypervigilance, avoidance, narrowed and rigid cognition about self and others, emotional numbing, social withdrawal, and cognitive inflexibility. In the language of the predictive-processing literature, the trauma produces overweighted priors — beliefs about danger and untrustworthiness that lock into place and resist updating. The world becomes smaller. Curiosity drops. Learning slows. The capacity to take in new information is diminished.
The cortical regions that thickened in the Stanford cohort — medial and lateral orbitofrontal, lateral occipital, entorhinal, lingual, middle and superior temporal, inferior parietal, pars orbitalis and triangularis — are not random. They are concentrated in the circuits the brain uses for flexible cognition, social inference, semantic integration, and the construction of new meaning from experience. The pattern is what the REBUS framework would predict: a transient relaxation of overweighted priors, followed by a reorganization in the direction of openness rather than rigidity.7
Recovery from trauma in this kind of intervention — on the broader psychedelic-assisted-therapy literature and consistent with what the Stanford clinical follow-up describes — looks less like symptom suppression and more like an inversion of the trauma phenotype. More curiosity. More willingness to take in new ideas. More social engagement. Restored capacity to update long-held beliefs about self and others. It is not the inverse of damage by subtraction. It is the inverse by addition — of the same flexibility, openness, and learning capacity the trauma had been actively keeping shut off, inaccessible.
This is what makes the longevity reading more than a metaphor. The phenotype that healthy aging at its best preserves — cognitive flexibility, social engagement, the capacity to learn and update — is the same axis trauma damages and the same axis the Stanford finding suggests this kind of intervention restores. A longer life, on that reading, is not the goal. A longer life that remains open, curious, social, and capable of remodeling itself is. The architecture that produces that — substrate-level plasticity, cortical thickening in flexibility-related circuits, neurotrophic-factor upregulation that movement and sleep and fasting also engage — is what the developmental-input reclassification is actually pointing at.
The category these compounds belong to is not the drug category. It is not, on its own, a longevity category either. It is a regeneration category. And what regenerates is not only tissue but the capacity for the kind of vibrant, thriving life that regeneration best serves.
A lineage older than the brain
The reason this kind of regeneration is even available — biologically, mechanistically — sits inside a lineage worth pausing on, because it changes how strange the result actually is.
Although we might associate intelligence and cognition with neurons, serotonin and its receptors are far older. The 5-HT receptors are G-protein-coupled receptors whose structural ancestors trace to early eukaryotes — single-celled organisms doing organizing work hundreds of millions of years before metazoan nervous systems existed.8 What kind of organizing work? The regulation of metabolic state, cellular response to environment, the maintenance of internal order against the entropic drift that all living systems exist within. Serotonin is, in this longer view, one of the molecules life uses to keep complexity from dissolving back toward energetic equilibrium. Away from entropy and chaos. When the complex brain finally emerged in octopuses, mammals, dogs, chimpanzees, whales, birds, and so many other kinds of life on Earth, it inherited the signaling system. The receptors did not have to be invented. They were repurposed.
This is the deeper claim Paul Stamets has been gesturing at when he describes psilocybin and the broader psychedelic class as something closer to a nutrient than a drug. The molecules and the receptors co-evolved in a thermodynamic context that predates everything we associate with mind. The fit between molecule and receptor is not a pharmacological accident. It is what very deep evolutionary contact produces.
The modern theory of how these compounds work fits the lineage. Robin Carhart-Harris and Karl Friston have argued — in Pharmacological Reviews — that psychedelics produce their reorganizing effects by transiently raising the entropy of cortical activity, relaxing the priors and predictive structures the brain has otherwise locked into. The framework is called REBUS, for Relaxed Beliefs Under Psychedelics. Read across the lineage, REBUS is a continuation of what these receptors have always done: modulate the relationship between a system and its entropy. What is new is the substrate. We have a cortex now, with belief structures and predictive models — the architecture of thought itself. The molecule is doing what serotonin’s receptors have done for hundreds of millions of years, but now to the structure that does the thinking.
Ibogaine — an indole alkaloid that sits adjacent to this tryptamine receptor lineage and engages it more peripherally than psilocybin or LSD — is part of the same evolutionary picture. The cortical thickening and brain-age reduction the Stanford team measured are what this very old reorganizing capacity, fully engaged, looks like in the modern substrate, in a damaged mind needing to regenerate in order to become whole again.
The window, and what fills it
There’s one more thing the structural data invites, and it’s the thing that to me makes the developmental-input framing personally actionable rather than only architectural.
A plasticity window is a temporary period during which the nervous system is more responsive than usual to inputs. New connections form more readily; priors update more easily; circuits restructure with less friction. The literature on psychedelic-induced plasticity — Gül Dölen’s work on reopening social-reward critical periods, the Carhart-Harris and Friston REBUS framework, the broader animal and human evidence on neurotrophic-factor upregulation — converges on the idea that compounds in this class open such windows.9 Ibogaine, on the Stanford evidence, appears to open a particularly large and durable one.
What goes through that window matters as much as the fact of it being open. The brain reorganizes around the inputs available during the period of heightened plasticity. Sleep architecture during those weeks. The consistency and quality of movement. The nutritional and inflammatory environment. The relational density of the people the recovering brain is in contact with. The autonomic state — sympathetic-dominant or recovery-capable — the nervous system spends its time in. These are not parallel “wellness practices” competing with the molecule for credit. They are the substrate the window builds with.
The framing extends beyond the brain. Preclinical work on ibogaine and its analogs suggests broad neuroimmune effects — modulation of microglia, peripheral immune signaling, the gut-immune axis, and the inflammatory tone of multiple tissues. The plasticity window appears to be partly a neuroimmune window, and the body’s tissues — not just the cortex — participate in the reorganization. Whatever modulates the body’s inflammatory and energetic state in the days and weeks around the dose plausibly modulates the magnitude of the result.
This is what the Adaptive Resilience domains are for. Each of the eight domains of Adaptive Resilience — sleep, movement, nutrition, hormesis and adaptation, nervous system regulation, environment, mental and cognitive growth, and social and collective alignment — names a class of input the body uses to maintain or rebuild its substrate. Each is a candidate determinant of how much of any given plasticity window translates into durable change. The compounds are not the whole story. They open the door, and the domains determine what the room becomes once you cross to the other side.
Of those domains, one is foundational in the sense that it conditions every other. It is also the one in which the variable most people optimize for is not the variable the most rigorous mortality data identifies. The next entry I’m writing is about that domain — and about why the right variable changes how almost everything else in the architecture interacts.
What this opens
We do not yet have the actual taxonomy of compounds the nervous system uses for structural reorganization. We have entries in it: psilocybin, ibogaine, ketamine, MDMA, perhaps DMT and 5-MeO-DMT, perhaps several entries we have not yet recognized. We have early mechanism data on each. We have, in ibogaine, the first human structural-MRI evidence that one of these compounds, given once with appropriate support, is associated with cortical thickening, subcortical expansion, and reduced biological brain age.
What we do not yet know is what distinguishes the compounds the brain uses for genuine remodeling from the compounds that merely modulate it. Which substrate states make each member of this class safe versus dangerous. Which conditions of trauma, rigidity, or accelerated aging each is best suited to address. Whether the brain-age reduction observed at one month persists, deepens, or attenuates over longer follow-up, in larger and more diverse populations.
That understanding is what the Stanford program — work that Adamson and the rest of the team are continuing in the wake of Dr. Nolan Williams’s death — is pushing the field toward. It is, to me, the most interesting frontier in this corner of medicine right now: not because of any single molecule, but because of what the convergence between Stamets, the receptor-evolution literature, and the new structural data is suggesting about the architecture of the nervous system itself, and about what a richer category of inputs to flourishing might look like.
The brain has a remodeling capacity. It uses signals — some endogenous, some inherited from a signaling architecture older than nervous systems — to engage that capacity. The receptors that bind these molecules predate the cortex by an unimaginable span of time. What is new is that we now have a cortex sophisticated enough that the reorganization is visible to us as cortical thickening, as subcortical expansion, as a younger predicted brain age, and, downstream, as changes in mood, in cognition, in the experience of being a self.
The work I’m doing is the framework that asks what the full taxonomy of inputs to flourishing looks like — not just psychedelic compounds, but movement, sleep, fasting, social contact, every substrate-level signal the nervous and metabolic systems use to organize themselves. Ibogaine is one entry, and the new Stanford data is one of the strongest pieces of evidence yet that the entry belongs on this newer model rather than the older one.
The next question is which others belong, and what the model’s structure turns out to be.
That question is the work, and the next entry is what I’m writing toward.
Notes on sources and framing
The Stanford clinical results10 and structural results come from the same observational cohort: thirty special operations veterans with blast TBI, single-dose oral ibogaine with IV magnesium pre-treatment. Both papers are peer-reviewed. The trial is open-label, lacks a placebo arm, and is small (n = 22 at one-month structural follow-up). The findings are striking and they license serious investigation; they do not yet license treatment recommendations.
The brainageR algorithm, like all machine-learning brain-age estimators, has known limitations: it is partly opaque mechanistically, and increases in cortical thickness in the cohort likely contributed to the lower age estimates. The 1.3-year reduction is a real finding within those constraints, not a clean biological readout.
The substrate-state framing of ibogaine cardiac risk — that risk is conditional on mitochondrial and electrolyte reserve, and that the historical record sampled the compromised end of that distribution — is a hypothesis consistent with the data but not directly tested by a controlled trial. I have flagged it as hypothesis throughout.
The receptor-evolution framing — that serotonin and 5-HT receptors predate nervous systems and that psychedelic compounds engage a reorganizing capacity older than the cortex — is well-grounded in the comparative biology and receptor-phylogeny literature. The synthesis with the entropic-brain / REBUS framework, and the further synthesis between this lineage and a longevity-coded reading of the Stanford structural results, is more recent and more contested.
The “developmental input” reclassification of psychedelics, and the longevity-adjacent reading of brain-age reduction in this cohort, are frontier framings. I find them productive. I have not represented them as established science.
This work is dedicated, in the iScience paper itself, to the memory of Dr. Nolan Williams.
Cherian KN, Keynan JN, Anker L, et al. Magnesium–ibogaine therapy in veterans with traumatic brain injuries. Nat Med. 2024;30(2):373–381. doi:10.1038/s41591-023-02705-w
Clinical outcomes: PTSD, depression, anxiety, functional disability; n=30 special operations veterans with blast TBI.
Geoly AD, Coetzee JP, Buchanan DM, et al. Increased cortical thickness and decreased brain age among special operations veterans with blast TBI after a magnesium-ibogaine protocol. iScience. 2026;29(3):115121. doi:10.1016/j.isci.2026.115121
Primary structural MRI data: cortical thickening, subcortical expansion, 1.3-year brain-age reduction; same cohort as Cherian 2024.
Cole JH, Poudel RPK, Tsagkrasoulis D, et al. Predicting brain age with deep learning from raw imaging data results in a reliable and heritable biomarker. NeuroImage. 2017;163:115–124. doi:10.1016/j.neuroimage.2017.07.059
brainageR algorithm: basis for predicted brain-age metric used in Geoly 2026.
Marton S, González B, Rodríguez-Bottero S, et al. Ibogaine administration modifies GDNF and BDNF expression in brain regions involved in mesocorticolimbic and nigral dopaminergic circuits. Front Pharmacol. 2019;10:193. doi:10.3389/fphar.2019.00193
GDNF/BDNF upregulation as proposed mechanism for ibogaine’s structural effects.
He DY, McGough NN, Ravindranathan A, et al. Glial cell line-derived neurotrophic factor mediates the desirable actions of the anti-addiction drug ibogaine against alcohol consumption. J Neurosci. 2005;25(3):619–628. doi:10.1523/JNEUROSCI.3959-04.2005
GDNF as primary mediator of ibogaine’s anti-addiction effects in preclinical model.
Azmitia EC. Modern views on an ancient chemical: serotonin effects on cell proliferation, maturation, and apoptosis. Brain Res Bull. 2001;56(5):413–424. doi:10.1016/S0361-9230(01)00614-1
Serotonin’s pre-neuronal evolutionary history; 5-HT receptor deep phylogeny.
Stamets P. Public lectures and writings on psilocybin, fungi, and mammalian neurobiology, 2017–2024.
Stamets stack and convergence hypothesis; no single primary source — refer to published interviews and lectures.
Carhart-Harris RL, Friston KJ. REBUS and the anarchic brain: toward a unified model of the brain action of psychedelics. Pharmacol Rev. 2019;71(3):316–344. doi:10.1124/pr.118.017160
REBUS framework: psychedelics relax priors, raise cortical entropy, enable reorganization of predictive structures.
Carhart-Harris RL, Leech R, Hellyer PJ, et al. The entropic brain: a theory of conscious states informed by neuroimaging research with psychedelic drugs. Front Hum Neurosci. 2014;8:20. doi:10.3389/fnhum.2014.00020
Foundational entropic-brain model; precursor to REBUS.
Nardou R, Sawyer E, Song YJ, et al. Psychedelics reopen the social reward learning critical period. Nature. 2023;618:790–798. doi:10.1038/s41586-023-06204-3
Dölen lab: critical-period reopening mechanism; MDMA restores social reward plasticity window.